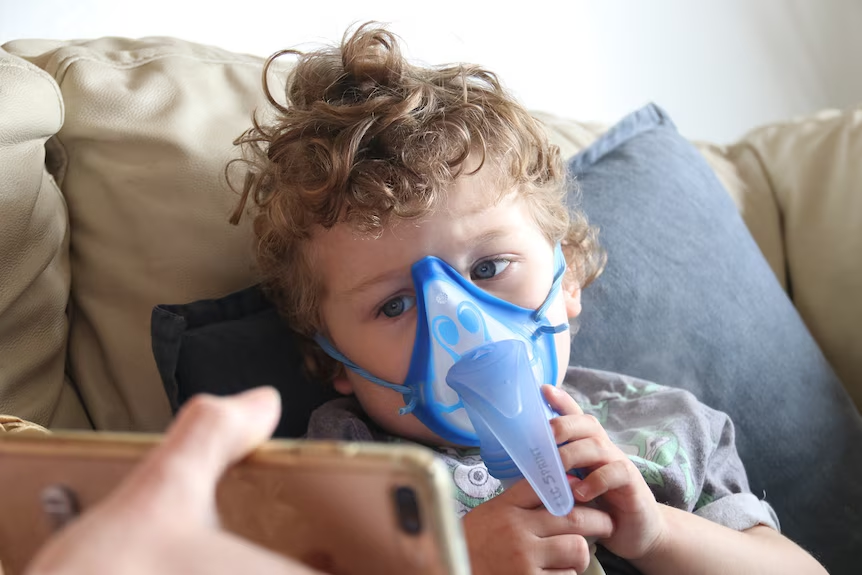
For the first time, scientists have mapped the earliest signs of lung damage in children with cystic fibrosis, revealing how the disease takes hold long before symptoms appear. The landmark study, led by researchers at the Murdoch Children’s Research Institute and the Peter MacCallum Cancer Centre, has produced the most detailed lung atlas ever created for young cystic fibrosis patients. The findings could transform how clinicians monitor and treat the condition, offering new hope for preventing irreversible lung damage that typically develops later in life. Cystic fibrosis is a genetic disorder that affects the lungs and digestive system, leading to chronic infections and progressive respiratory decline. While advances in treatment have extended life expectancy, early lung damage often goes undetected until it becomes severe. This new research provides a critical window into the disease’s earliest stages, enabling clinicians to intervene sooner and potentially alter its long term trajectory.
Clinical Significance
Cystic fibrosis is a life limiting genetic condition that affects more than 70,000 people worldwide. Despite significant improvements in care, lung disease remains the leading cause of morbidity and mortality in these patients. The new lung atlas offers an unprecedented look at how the disease progresses at a cellular level, even in very young children. This level of detail could help clinicians identify biomarkers for early intervention, tailor treatments to individual patients, and ultimately slow or prevent the irreversible lung damage that defines the later stages of the disease.
Deep Dive and Research Findings
The study analyzed lung samples from young children with cystic fibrosis, creating a comprehensive map of the lower airways. Using advanced imaging and molecular techniques, the research team identified specific patterns of inflammation, mucus buildup, and structural changes that occur long before traditional diagnostic tools like spirometry can detect abnormalities. These findings challenge the long held assumption that lung damage in cystic fibrosis begins only after symptoms appear, suggesting instead that the disease’s roots may be present from infancy.
The lung atlas also revealed variations in how different regions of the lungs are affected, providing clues about why some patients experience more rapid disease progression than others. For example, areas with higher mucus concentration showed greater structural damage, while regions with less inflammation appeared relatively preserved. This spatial understanding of disease progression could inform more targeted therapies, such as inhaled medications designed to reach the most vulnerable parts of the lungs.
Future Outlook and Medical Implications
The implications of this research extend far beyond the laboratory. By identifying the earliest signs of lung damage, clinicians may soon be able to implement personalized treatment plans that address the unique progression of the disease in each patient. For instance, children identified as high risk for rapid lung decline could receive more aggressive therapies, such as CFTR modulators or anti inflammatory drugs, before irreversible damage occurs.
The study also opens the door to new research avenues, including the development of non invasive diagnostic tools that could detect early lung changes without the need for invasive procedures. Additionally, the lung atlas could serve as a benchmark for evaluating the effectiveness of emerging therapies, helping researchers determine whether new treatments are truly halting disease progression or merely masking symptoms.
Patient or Practitioner Guidance
For parents of children with cystic fibrosis, this research underscores the importance of early and consistent monitoring. While the disease is often asymptomatic in its earliest stages, regular check ups that include advanced imaging or biomarker testing could help detect subtle changes in lung health. Parents should discuss with their child’s healthcare team whether emerging diagnostic tools, such as those used in this study, are available or recommended for their situation.
For clinicians, the findings highlight the need to move beyond traditional lung function tests, which may not capture early disease activity. Incorporating molecular or imaging based assessments into routine care could provide a more accurate picture of a patient’s lung health and guide treatment decisions. The study also reinforces the value of multidisciplinary care, as early intervention may require input from pulmonologists, geneticists, and respiratory therapists to address the complex needs of young cystic fibrosis patients.
While the lung atlas is a significant step forward, researchers caution that more work is needed to translate these findings into clinical practice. Future studies will likely focus on validating the biomarkers identified in the atlas and determining how best to integrate them into existing care protocols. In the meantime, the research offers a hopeful glimpse into a future where cystic fibrosis is managed proactively, rather than reactively.
Key Takeaways
- The largest lung atlas of young cystic fibrosis patients reveals early signs of lung damage, even before symptoms appear.
- Advanced imaging and molecular techniques identified specific patterns of inflammation and structural changes in the lower airways.
- The findings could lead to earlier, more targeted interventions to prevent irreversible lung damage in cystic fibrosis patients.
- Clinicians may soon use personalized treatment plans based on individual disease progression, improving long term outcomes.
- Parents and practitioners should prioritize early monitoring and discuss advanced diagnostic tools with their healthcare teams.
Frequently Asked Questions
What is cystic fibrosis, and how does it affect the lungs?
Cystic fibrosis is a genetic disorder that causes thick, sticky mucus to build up in the lungs and other organs. In the lungs, this mucus clogs airways and traps bacteria, leading to chronic infections, inflammation, and progressive lung damage over time.
Why is this new lung atlas significant for cystic fibrosis care?
The lung atlas provides the most detailed map yet of how cystic fibrosis affects the lungs in young children. By identifying early signs of damage, it could help clinicians intervene sooner, potentially preventing or slowing the progression of lung disease.
How might this research change the way cystic fibrosis is treated?
The findings could lead to more personalized treatment plans, where therapies are tailored to the specific patterns of lung damage in each patient. This might include earlier use of CFTR modulators, anti inflammatory drugs, or other targeted interventions.
What should parents of children with cystic fibrosis do with this information?
Parents should discuss the potential benefits of advanced monitoring tools with their child’s healthcare team. Early detection of lung changes could allow for timely interventions, so staying informed about emerging diagnostic options is important.
Are there any new treatments on the horizon for cystic fibrosis?
While this study does not introduce a new treatment, it provides a foundation for developing more precise therapies. Researchers are continually exploring new drugs, gene therapies, and other interventions to improve outcomes for cystic fibrosis patients.
Medical Review: MedSense Editorial Board







DISCUSSION (0)
POST A COMMENT